INTRODUCCIÓN
El xeroderma de pigmentoso (XP) es una afección genética caracterizada por una extrema sensibilidad a la radiación ultravioleta, como la luz del sol. También es llamado melanosis lenticular progresiva. Es una entidad trasmitida por herencia autosómica recesiva y cuyo cuadro clínico es un reflejo de la alteración en la reparación del ADN dañado por radiación ultravioleta (RUV). Ocurre en ambos sexos y en todas las razas. (1,2) Quienes lo padecen suelen quemarse fácilmente con el sol y a corta edad muestran daño actínico severo con presencia de pecas y otras manchas pigmentadas, así como xerosis, de aquí el nombre de la enfermedad. Aún en la infancia temprana comienzan a aparecer queratosis y queilitis actínicas y finalmente lesiones malignas cutáneas: carcinoma basocelular, espino celular o melanoma maligno. Además, es frecuente en estos enfermos la presencia de carcinomas epidermoides en mucosa oral, tumores pulmonares, renales, testiculares, gástricos, pancreáticos, mamarios, uterinos y cerebrales, así como leucemias. (2-4)
Hasta el 30 % de los enfermos con XP presenta alteraciones neurológicas, que comprenden desde una hiporreflexia casi imperceptible, hasta crisis convulsivas, espasticidad generalizada, retardo mental o sordera sensorial. A nivel oftalmológico, puede ocasionar queratitis y opacidad cornial con neovascularización. (1, 3, 5)
Los enfermos con XP tienen un defecto a nivel de la endonucleasa, en la reparación del ADN dañado por RUV (y en menor grado otros agentes dañinos como fármacos y carcinógenos químicos). Con ello se van acumulando y perpetuando mutaciones en el ADN que igualmente se reflejan en el desarrollo de una neoplasia maligna. La célula de algunos enfermos presenta rupturas cromosómicas, existe heterogeneidad genética en cuanto al sitio específico de los defectos que no pueden ser reparados por ciertos enfermos y a estos se les llama grupos complementarios. Se han descrito por lo menos 8 grupos complementarios diferentes en enfermos con XP. (A, B, C, D, E, F, G y variante), aunque el cuadro clínico y prematuro de la enfermedad suelen ser el mismo, el grado de diferencia en la reparación del ADN es muy diferente entre ellos (de 2 % a 100 % de lo normal). (3, 6)
La forma más grave del XP es la del de Santis- Cacchione que se caracteriza por trastornos oculares, neuropsiquiátricos, endocrinos y óseos. (7)
Se plantea que el síntoma más frecuente es la fotofobia, (5,8) mientras que al examen físico pueden observarse quemadura solar y ampollas, después de una exposición mínima al sol que no sana, telangiectasia cutánea, incremento de la pigmentación regular de la piel, formación de costras, descamación, exulceraciones y exudación, así como cambios neurológicos.
El examen ocular puede revelar opacidad de la cornea, queratitis, tumores en el párpado y blefaritis.
El XP se observa con frecuencia cuando hay consanguinidad, por lo que es importante el diagnóstico prenatal y prevenir las posibles complicaciones que se puedan presentar.
El diagnóstico prenatal es posible por la prueba de vellosidad coriónica y cultivo de las células amnióticas; después del nacimiento, es posible mediante cultivo de los fibroblastos de la piel y por biopsia de la piel.
Las complicaciones pueden ser de diversa índole: desfiguramiento, tumores de las células basales, tumores de las células escamosas y melanoma maligno. (7, 8)
Los diagnósticos diferenciales principales son otras dermatosis inducidas o agravadas por la luz, o enfermedades hereditarias con aumento de la hipersensibilidad celular: síndrome de Bloom, síndrome de Rothmun-Thompson y progeria.
El tratamiento se basa en vitaminas antioxidantes, analgésicos, antibióticos y bloqueador solar, este con factor 30 o más, además de tratamiento quirúrgico en caso de carcinoma o melanoma. Los pacientes deben utilizar anteojos muy oscuros como protección contra la luz ultravioleta. (8)
PRESENTACIÓN DEL CASO
Paciente YGS de 12 años de edad, piel blanca, femenina, de procedencia rural, con antecedentes de padecer desde los 8 meses de edad, y luego de una exposición al sol, de “quemaduras con ampollas” de +- 3cm en cara y tronco. El médico de familia y el pediatra del área le diagnosticaron dermatitis solar y le orientaron a la madre la no exposición solar. Fue valorada por el dermatólogo del área a los 6 años de edad, el cual le diagnosticó xeroderma pigmentoso. A los 10 años se le realizó biopsia de piel, confirmándose el diagnóstico. Se remitió al centro de Referencia Nacional donde se mantiene bajo control y seguimiento.
Antecedente patológicos personales: Xeroderma pigmentoso, apendicetomía.
Antecedentes patológicos familiares: Abuela paterna: hipertensión arterial; abuela materna: cardiopatía isquémica; tíos paternos: piel actínica.
Examen físico: fotofobia.
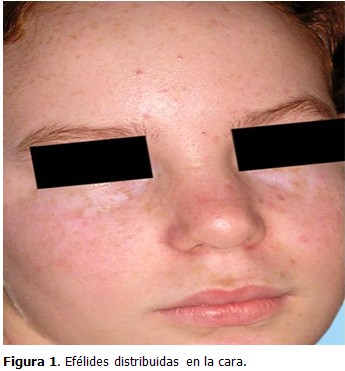
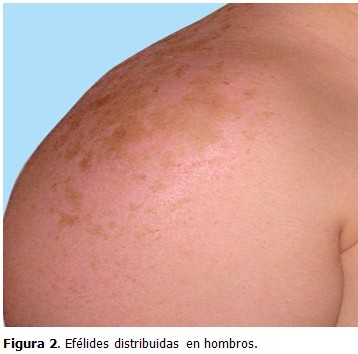
Examen dermatológico: piel tipo I, múltiples efélides distribuidas en cara (dorso de nariz, mejillas, frente) y espalda. (Figuras 1 y 2)
Examen oftalmológico: queratitis
Examen psicológico: normal. Coeficiente intelectual promedio.
Exámenes complementarios: hemoglobina: 12,4 g/L; hematocrito: 0,43; leucograma: leucocitos 7,3 x 109/L; stab: 000; segmentados: 049; eosinófilos: 003; linfocitos: 048; monocitos: 000; eritro: 20 mmol/H; glicemia: 4,3 mmol/L.
Resultado de la dermotopatología:
Biopsia de piel que muestra por hematoxilina y eosina: hiperqueratosis sin paraqueratosis, adelgazamiento de la capa de Malpighio, sin prolongaciones interpapilares, dándole un aspecto atrófico a la epidermis, pigmentación melánica focal de la capa basal y algunos melanóforos en el corion superior, edema de la dermis superior y degeneración del colágeno a este nivel evidenciado por técnica tricómica de Prico- Mallory.
Conclusión: xeroderma pigmentoso.
La paciente evolucionó sin complicaciones. El tratamiento estuvo conformado por consejos genéticos, medidas generales (protección solar con factor de protección al 30 %) e indicación de algunos medicamentos (vitaminas antioxidantes, analgésicos, cremas hidratantes y antibióticos en caso de necesidad).
DISCUSIÓN
La incidencia mundial de la enfermedad es de 2 a 4 nacidos vivos por millón. La enfermedad varía de un país a otro, debido a aspectos étnicos, geográficos y costumbres de ciertas regiones como casamientos entre familiares. (6,7)
La frecuencia con que se da esta enfermedad en los Estados Unidos de América y Europa, es de 1:250 000. En Japón, la tasa asciende a razón de 1:40 000 y el grupo XPA es el más frecuente, mientras que en Estados Unidos es el XPC. Una incidencia alta ha sido reportada en Libia, Arabia y Egipto. En Brasil, de 1953 a 1995, se informaron 48 pacientes con XP, quince de ellos fueron seguidos en su enfermedad; la consanguinidad estuvo presente en ocho de ellos. (6,7) En Cuba se han diagnosticado 63 casos, en Cienfuegos hasta el momento hay 5 casos diagnosticados. (8)
No existen evidencias de preferencia por una u otra raza, ni entre hembras y varones. (6-8)
En lo referente a la mortalidad, los individuos con esta enfermedad desarrollan neoplasias cutáneas múltiples en una edad joven. Dos causas importantes de la mortalidad son el melanoma metastático y el carcinoma escamoso. Los pacientes de edad menor a 20 años tienen un aumento en la incidencia del cáncer y del cáncer no melanoma. La edad media del cáncer de la piel es de 8 años en los pacientes con XP, frente a 60 años en la población normal. Los primeros síntomas ocurren durante los primeros dos años de vida. (7,8)
Los pacientes con XP son también susceptibles a la infección y, en algunos subtipos, a las complicaciones neurológicas. Menos del 40 % de los pacientes sobrevive más allá de la edad de 20 años. Los individuos con una enfermedad con poca expresividad pueden sobrevivir más allá de la edad media. (6,8)
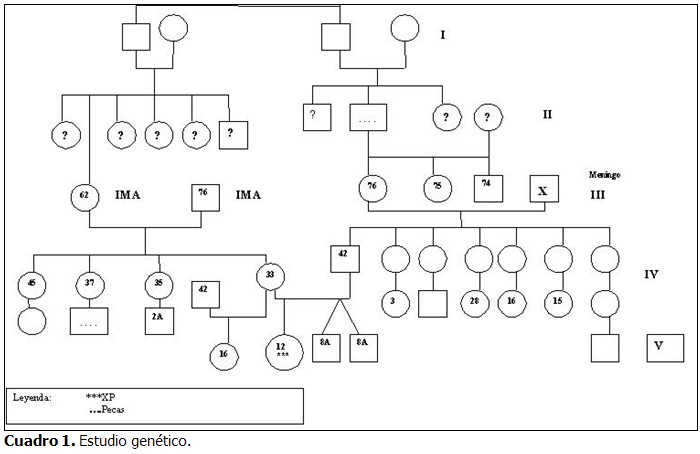
En el caso presentado, el diagnóstico de xeroderma pigmentoso no se vio relacionado con los antecedentes familiares, sin embargo, el estudio genético (Cuadro 1) reveló consanguinidad.