INTRODUCCIÓN
El raquitismo fue descrito por primera vez en el siglo XVII por los ingleses Daniel Whistler y Francis Gilsson. Estos estudiaron una gran familia de Carolina del Norte, con raquitismo resistente a la vitamina D y en él reconocieron que la hipofosfatemia fue el marcador más relacionado con esta enfermedad, por lo tanto surgió el término "raquitismo hipofosfatémico resistente a la vitamina D".1-3
El raquitismo hipofosfatémico (RH) es una enfermedad que se incluye dentro de un grupo de enfermedades hereditarias caracterizadas por la pérdida de fosfato por los riñones, que ocasionan retardo del crecimiento, raquitismo y osteomalacia. La forma más común es el raquitismo hipofosfatémico dominante ligado al cromosoma X, el cual es causado por mutaciones en el gen PHEX, con penetrancia completa después del año de edad, se ha localizado en el locus Xp22.1. Su incidencia se estima de 1: 20 000 nacidos vivos.3
Las otras formas de los síndromes hipofosfatémicos hereditarios presentan menor prevalencia. Estas incluyen el raquitismo hipofosfatémico autosómico dominante, el raquitismo hipofosfatémico autosómico recesivo tipos 1 y 2 y el raquitismo hipofosfatémico hereditario con hipercalciuria.4
El raquitismo hipofosfatémico autosómico dominante (ADHR) está causado por mutaciones de ganancia de función en FGF23 que impiden su escisión proteolítica; el factor de crecimiento de fibroblastos 23 (FGF23) es una hormona que inhibe la reabsorción renal de fosfato y la biosíntesis de 1,25-dihidroxivitamina D.4 Las concentraciones bajas de hierro juegan un papel en la fisiopatología de ADHR. La deficiencia de hierro es un desencadenante ambiental que estimula la expresión de FGF23 y la hipofosfatemia en DADDH. Varias mutaciones han sido recientemente identificadas en los genes PHEX, FGF23, Dmp1 y ENPP1 en pacientes con HR.5
La principal característica de la enfermedad es una pérdida inadecuada de fosfato a través de los riñones, lo que conduce a altas concentraciones de fosfato en la orina (fosfaturia) e hipofosfatemia con la mineralización ósea defectuosa. Los pacientes tienen raquitismo y osteomalacia, deformidades en las extremidades inferiores, dolor de huesos, baja estatura, anormalidades dentales y alteración del metabolismo de la vitamina D.5-7
Por ser una enfermedad hereditaria poco común y tener implicación ósea, así como considerar valioso el que se realice un diagnóstico oportuno que beneficie el seguimiento y tratamiento en equipo multidisciplinario, que permita, además, brindar un adecuado asesoramiento genético a los familiares, se decidió la presentación del caso.
PRESENTACIÓN DEL CASO
Se presenta el caso de una paciente de tres años de edad, quien después de ser atendida en consulta de Ortopedia y de Endocrinología, fue remitida a consulta de Genética Clínica, por presentar deformidades en miembros inferiores, en especial la articulación de la rodilla.
Hija de padres sin defectos congénitos visibles al examen físico, no consanguíneos, en edades de bajo riesgo genético, con antecedentes de salud.
Antecedentes prenatales: ligera anemia en el segundo trimestre del embarazo.
Antecedentes perinatales: nace por parto distócico, inducción fallida, por lo que se practicó cesárea a las 41,3 semanas; peso 3210 gramos; talla 49 cm; Apgar 8-9.
Antecedentes posnatales: primer año de vida con buena ganancia de peso y buen desarrollo psicomotor.
Alrededor de los 19 meses la madre comenzó a observar una ligera deformidad de las piernas que se fue acentuando con los días.
Al examen físico se observó genu varum simétrico que determina una marcha «anadeante». (Figura 1).
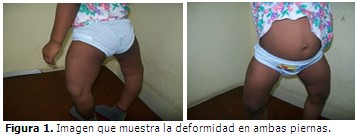
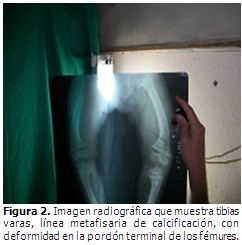
Esta deformidad de ambas piernas se confirmó radiológicamente por la presencia de: tibias varas, línea metafisaria de calcificación presenta apariencia irregular y "deshilachada", conformando la imagen "en copa" con concavidad hacia el lado epifisario, produciendo deformidad en los fémures. (Figura 2).
Los exámenes complementarios mostraron los siguientes resultados: glucemia 5,3 mmol/L; creatinina 53 mmol/L; calcio sérico 2,08 mmol/L; fosfatasa ácida 6,83 mmol/L; fosfato sérico 0,6 mmol/L; Hb 11,6 g/l; plaquetas 273 X 109/L; eritro 8 mml/L
El perfil bioquímico sérico determinó la existencia de niveles normales de calcio, con una hipofosfatemia severa, confirmándose la sospecha clínica de que la paciente tiene un raquitismo hipofosfatémico autosómico dominante, el cual tiene penetrancia reducida y expresividad variable.
DISCUSIÓN
El término de raquitismo hipofosfatémico abarca un conjunto de síndromes genéticos caracterizados por: afectación de la mineralización esquelética, hiperfosfaturia e hipofosfatemia, en ausencia de deficiencia de vitamina D. Este cuadro clínico es causado por un incremento de los niveles del factor de crecimiento fibroblástico (FGF)-23, que, actuando sobre su receptor, impide una reabsorción adecuada del fosfato en el túbulo renal proximal e interfiere en la hidroxilación renal de la vitamina D.1,2 Para mantener un balance neutral del fosfato, la cantidad de fosfato absorbida en el intestino debe ser semejante a la cantidad excretada, principalmente en la orina. Este balance es mantenido y regulado por la acción coordinada de diferentes hormonas y factores, como la hormona paratiroidea, el factor de crecimiento fibroblástico-23 (FGF-23) y la forma activa de la vitamina D, la 1α,25-dihidroxivitamina D3 (1α,25(OH)2D3).3
En los niños suele manifestarse cuando comienzan a caminar, con arqueamiento de las piernas y otras deformidades óseas, seudofracturas, dolor óseo y talla baja. Las excrecencias óseas en las inserciones musculares pueden limitar el movimiento, como lo que sucede en esta paciente con limitación en los movimientos de la rodilla.3
El raquitismo hipofosfatémico autosómico dominante (RHAD) se caracteriza por la presencia de pérdida renal de fosfatos con desarrollo de hipofosfatemia persistente en familias con la forma autosómica dominante; el gen implicado FGF23 se encuentra en el cromosoma 12p13.4
El RH es una enfermedad que se produce por trastorno de transporte de fosfato, dentro de estos se describe la hipofosfatemia familiar. Se describen varias formas hereditarias dentro de los cuales está el patrón de herencia autosómico dominante, autosómico recesivo, recesivo y dominante ligado al cromosoma X, también los casos esporádicos relacionados con raquitismo oncógeno.4 Por último, está el raquitismo seudocarencial, dentro de estos el tipo 1, por trastorno de la L-hidroxilasa, el tipo 2, por trastorno de receptores de la 1,25(OH) 2D3, así como las hipercalciurias idiopáticas. Incluso, dentro de este subgrupo existe una notable variabilidad clínica y evolutiva, con respuesta heterogénea al tratamiento.5
La principal característica de la enfermedad es una pérdida inadecuada de fosfato por vía renal, lo que conduce a una alta fosfaturia e hipofosfatemia con la mineralización ósea defectuosa. Los pacientes tienen raquitismo y osteomalacia, deformidades en las extremidades inferiores, dolor de huesos, baja estatura, anormalidades dentales y alteración del metabolismo de la vitamina D. El diagnóstico se realiza por las concentraciones séricas de fosfato, fosfatasa alcalina y 1,25-dihidroxivitamina D3.6 El tratamiento consiste en la administración oral de fosfato más calcitriol.
Los medicamentos para la terapia de esta enfermedad pueden conducir a complicaciones a largo plazo, tales como el hiperparatiroidismo secundario e insuficiencia renal. Por lo tanto, el tratamiento más seguro y eficaz es corregir el exceso del factor de crecimiento fibroblástico (FGF23), con anticuerpo anti-GF23. En estos momentos se utilizan métodos de biología molecular como los polimorfismos de simples nucleótidos, a través de la técnica de microarray para identificar mayor número de mutaciones en estos genes implicados, con el objetivo de realizar un tratamiento más individualizado para cada mutación.7-9 El actual tratamiento de los pacientes con cualquier tipo de hipofosfatemia consiste en la suplementación de fosfato y de formas bioactivas de vitamina D.
La heterogeneidad clínica y genética, tanto alélica como no alélica, de esta enfermedad dificulta la identificación del patrón de herencia, así como su clasificación clínica y el pronóstico de la evolución. El curso de la enfermedad y su respuesta tan variable al tratamiento responden a cambios epigenéticos, aún no dilucidados completamente.10
Ante esta paciente femenina con manifestaciones clínicas sugestivas de un raquitismo que se comprobó a través de los estudios de la química sanguínea y después de analizados las radiografía de los miembros inferiores preferentemente de fémur, tibia y peroné donde se observaron signos característicos de esta enfermedad, que permitieron realizar el diagnóstico clínico de esta paciente.
