INTRODUCCIÓN
La poroqueratosis (PQ) es una dermatosis epidérmica que se caracteriza por lesiones anulares, queratósicas e hiperpigmentadas, únicas o múltiples, de evolución crónica; es una alteración específica de la queratinización y se considera como una lesión premaligna con riesgo elevado de producción de carcinoma epidermoide. Se han reconocido seis variantes clínicas de la enfermedad: la PQ de Mibelli clásica, la PQ superficial diseminada (actínica, no actínica y de inmunosuprimidos), la PQ lineal, PQ palmoplantar y diseminada y la PQ facial atípica y sindromática.1 Las lesiones de la PQ clásica fueron descritas por Mibelli, dermatólogo italiano, en 1893, quien les asignó el nombre de poroqueratosis, porque creyó que estas lesiones comenzaban en las glándulas sudoríparas.2 Una forma diseminada más superficial fue descrita por Respighi alrededor de ese mismo tiempo, y la variante lineal se incluye en este mismo siglo; en 1966 la PQ actínica superficial diseminada fue descrita por Chernosky y en 1971 Guss y colaboradores añadieron la palmoplantar diseminada.3,4
La coexistencia de varias formas de PQ en un mismo paciente, o en varios miembros de una familia, sugiere que está representado por un defecto genético común. Algunos autores incluyen otra forma clínica, denominada gigante, muy rara.5,6
Pueden aparecer múltiples lesiones, pero son casi siempre unilaterales y localizadas regionalmente. Las lesiones faciales de PQ son raras y existen pocos casos reportados. Algunas de ellas son destructivas y otras son lesiones no atróficas superficiales. El diagnóstico diferencial de debe realizarse con la psoriasis en placa, el nevus epidérmico lineal verrugoso y el nevus ostial ecrino poroqueratósico.7-9 El diagnóstico se realiza por la clínica, antecedentes familiares e histopatología.
El tratamiento es un desafío en todas las variantes de la PQ y depende del sitio y extensión de las lesiones. Las lesiones circunscriptas de la PQ de Mibelli lineal pueden ser resecadas y tratadas con injerto, electro cirugía y crioterapia. Además de la dermoabrasión se usan otras pautas terapéuticas, como queratolíticos tópicos, retinoides tópicos, el calcipotriol, el 5-fluoracilo, láser CO2, etretinato, acetretin e imiquimod.5,8 Otro tratamiento con buenos resultados ha sido la terapia fotodinámica en España.10
El presente artículo expone el tratamiento y evolución de un paciente con diagnóstico de PQ, enfermedad muy poco frecuente, tanto en Cuba como en el resto del mundo.
PRESENTACIÓN DEL CASO
Se presenta el caso de un paciente masculino, de 18 años de edad, de color de la piel blanco y procedencia rural. Fue el producto de un embarazo normal, parto eutócico a término, con buen peso y apgar al nacer, pero su madre refirió que a los cuatro meses de edad comenzó a presentar lesiones en la piel de las axilas, que luego se extendieron a los miembros superiores en forma lineal, diseminándose a la cara, tronco y miembros inferiores de color pardo oscuro y aspecto áspero a la palpación. Por la extensión de las lesiones y la falta de mejoría a pesar de los tratamientos indicados, fue remitido al Hospital Pediátrico Paquito González Cueto, de Cienfuegos.
Al ser evaluado en la consulta de Dermatología, con cuatro años de edad, la madre refirió como antecedentes patológicos familiares: asma bronquial (madre), diabetes mellitus (abuelo).
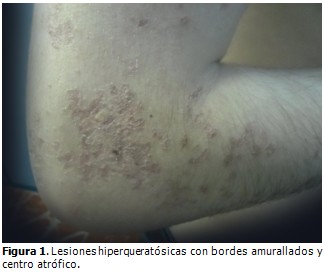
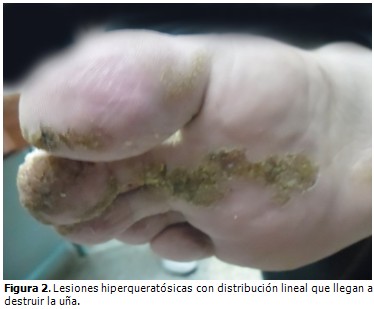
Al examen físico se observaron lesiones hiperqueratósicas, pardas y lineales, en región de las axilas, brazos, periné, muslos y piernas. En las lesiones de los brazos se observó un centro atrófico con bordes hiperqueratósicos; en la cara, pápulas hiperqueratósicas, al igual que en el resto de las regiones afectadas. En el miembro inferior derecho las lesiones se extendían hasta la planta del pie.
Se le indicaron varios exámenes complementarios, con los siguientes resultados: hemoglobina (hb): 12,3 g/l, hematocrito (hto): 0,38, leucocitos: 9,63x10/9, stab: 000, seg: 0,41, eosinófilos (eos): 006, monocitos (mon): 000, linfocitos (linfo):0,53.
Ultrasonido abdominal: hígado que rebasaba dos cm el reborde costal derecho con patrón homogéneo; vesícula, páncreas, riñones y bazo normales.
Intubación duodenal normal.
Citología de impresión conjuntival (CIC): severamente alterado, déficit de vitamina A.
Transaminasas (TGP): 7,3 unidades.
Fondo de ojo: sin alteraciones.
Biopsia de piel: (CR-03-516) evidenció hiperqueratosis, atrofia de la capa mucosa de Malpighi, fibrosis de la dermis superior con presencia de la lamella cornoide e infiltrado inflamatorio crónico. Se concluyó que se trataba de una PQ en su variante superficial diseminada.
Valoración genética clínica: al realizar estudio familiar, se pudo observar en el árbol genealógico que solo el paciente presentaba la enfermedad, por lo que se infirió que se trataba de una mutación de novo.
Examen psicológico: sin alteraciones en su desarrollo psíquico; coeficiente intelectual normal promedio.
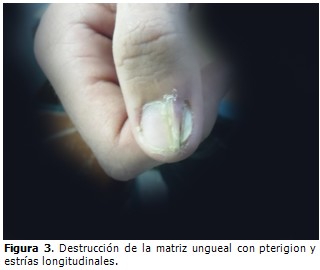
Evolución y estado actual del paciente: desde el año 2003 el paciente fue seguido en consulta de genodermatosis, llevando varios tratamientos médicos, tales como: uso de cremas emolientes, queratolíticos, protectores solares, vitaminoterapia, cremas esteroideas, acitretin sistémico (el cual provocó reacciones secundarias que motivaron la supresión del medicamento) y seguimiento por psicología. A pesar de esto las lesiones en piel han empeorado, generalizándose por todo el tegumento cutáneo, predominando el patrón lineal, aumentando en número y tamaño, llegando a ser grandes placas con centro atrófico deprimido, lampiño, anhidrótico hipo o hiperpigmentado, rodeado por bordes queratósicos ligeramente elevados, que generan un resalto al tacto, extendidas hasta el lecho ungueal de algunos dedos de los miembros superiores e inferiores, siendo evidentes la destrucción de la matriz ungueal, la opacidad de la lámina ungueal y las estrías longitudinales. (Figura 1, figura 2 y figura 3). A pesar de las diferentes terapias tópicas y sistémicas, no se logró estabilidad ni curación de la enfermedad.
DISCUSIÓN
La evolución del caso presentado fue similar a lo referido en la literatura. Es una enfermedad que comienza durante la infancia y las lesiones aumentan con los años, generalmente son asintomáticas. Los hombres son más afectados que las mujeres en una proporción de 3 a 1 y afecta a cualquier raza. Se conocen aproximadamente 250 casos en la bibliografía mundial. La forma lineal es más frecuente en los niños y adolescentes, la actínica aparece después de los 16 años y en la etapa adulta, y suele ser más frecuente en personas susceptibles (personas con VIH y receptores de trasplantes).6 Su cuadro clínico está caracterizado al inicio por lesiones de tamaño variable de unos pocos milímetros a varios centímetros, con un centro rozado, eritematoso, ligeramente atrófico y márgenes hiperqueratósicos elevados. En otros casos pueden ser carmelitas o pardas más oscuras, formando placas anulares, que hacen relieve con bordes bien delimitados hiperqueratósicos, por lo general de más de un milímetro de altura y contienen ranuras o surcos como un dique dividido. Las lesiones tienen forma crateriforme, localizadas fundamentalmente en regiones acrales de la extremidades, en los muslos y región peri genital, y más raramente afectan el lecho ungueal.9 Las lesiones del paciente presentado coinciden con muchas de estas características, dado su aspecto hiperqueratósico, su disposición amurallada, con distribución lineal y centro atrófico.
En todos los casos de PQ revisados, esta se considera como enfermedad genética autosómica dominante, lo cual está bien establecido debido a la presencia de clones celulares que muestran grados variables de displasia. Se ha demostrado inestabilidad en el brazo corto del cromosoma 3; la causa es desconocida, aunque se postulan varias hipótesis que indican que la epidermis y la dermis son las responsables de producir la enfermedad. Las células epidérmicas de la lesión poroqueratótica muestran un alto ritmo de ADN anormal, lo que sugiere que las lesiones tienen un mínimo potencial de malignidad.4 Al paciente se le realizó una valoración genética clínica, sin embargo, en su caso se trató de una mutación de novo, lo que aporta a la práctica dermatológica otra posible etiología.
Histológicamente se identificó la lamella cornoide, patrón descrito como característico de esta enfermedad en todos los casos publicados, caracterizado por la presencia de hiperqueratosis en su centro atrófico, atrofia de la capa mucosa de Malpighi y fibrosis de la dermis superior.1
Tal y como se ha evidenciado en este paciente, la PQ es una enfermedad de curso progresivo. El patrón clínico de sus lesiones, extendidas en el tegumento cutáneo llegando a provocar deformidades ungueales, es similar al de casos precedentes. A pesar de las diferentes terapias utilizadas no se logró estabilidad ni curación de la enfermedad.
