INTRODUCCIÓN
Las distrofias musculares son trastornos hereditarios que comprenden un espectro amplio de afectación muscular progresiva como consecuencia de distintas alteraciones en genes codificantes de las proteínas de la membrana celular, el citoesqueleto y el núcleo de los miocitos, por lo que el compromiso cardiaco de estas enfermedades es usual. Dentro del gran grupo de las enfermedades neuromusculares, la distrofia miotónica de Steiner tiene muy baja incidencia. Afecta con más frecuencia a los adultos, con un patrón genético hereditario de carácter autosómico dominante: alteraciones en el brazo largo del cromosoma 19 que codifica la miotoninaproteinsinasa, consistente en la repetición de un trinucleótido expandido en dicho cromosoma.1-3
La distrofia miotónica de Steinert es la distrofia muscular más frecuente después del Duchenne. Su incidencia según reportes de algunos países, es de 13,5 casos por 100 000 habitantes. Afecta más a los hombres que a las mujeres. Tiene un amplio espectro clínico, ya que afecta otros sistemas además del muscular; se manifiesta desde las formas neonatales más graves hasta las formas oligosintomáticas en adultos. Algunos de sus signos fundamentales son la distrofia muscular, lossignos miotónicos, calvicie precoz y progresiva, trastornos de la conducción y del ritmo cardiaco; puede estar asociada a diabetes (resistencia a la insulina), cataratas subcapsulares posteriores, atrofia gonadal, déficit intelectual y reducción de la motilidad esofágica y colónica, entre otros padecimientos.4
Las causas de insuficiencia cardiaca (IC) están generalmente en relación con las enfermedades cardiovasculares, que incluyen aterosclerosis coronaria, hipertensión arterial, las miocardiopatías, la diabetes mellitus,lesiones valvulares congénitas y adquiridas y otras malformaciones congénitas.Pero en los últimos años se ha enfatizado en las causas genéticas moleculares de las enfermedades miocárdicas. Las principales causas genéticas de la IC son las miocardiopatías familiares, las hemoglobinopatías, los trastornos mendelianos de la matriz extracelular y los trastornos neuromusculares y musculares.4
La electromiografía (EMG) pone de manifiesto el fenómeno miotónico, que excepto en las formas congénitas, se detecta antes de la aparición de manifestaciones clínicas. Lo que caracteriza y define a esta enfermedad, es precisamente la dificultad para relajar los músculos después de una contracción mantenida, lo que se denomina fenómeno miotónico. Los valores de las enzimas musculares séricas suelen ser normales o moderadamente elevados.La biopsia muscular muestra un elevado porcentaje de fibras con núcleos internos, fibras en anillo y atrofia de las fibras tipo 1. En la actualidad no suele practicarse, pues el diagnóstico se basa en la clínica, EMG y estudios genéticos. No se dispone de un tratamiento específico.1-8
El artículo puede ser de interés para todo profesional de salud, pero de manera particular para especialistas de Medicina Interna, Neurología y Medicina Familiar, así como para aquellos en formación de posgrado, ya que se expone el caso de una enfermedad poco frecuente.
PRESENTACIÓN DEL CASO
Se presenta el caso de un paciente de 47 años de edad, masculino, de color de piel blanco, que acudió al Servicio de Urgencias y Emergencias por falta de aire en horas de la noche, sin alivio. Esta falta de aire había comenzado un mes antes, al realizaresfuerzos medianos, siendo más aguda en horario nocturno, lo que le obligaba a permanecer sentir alguna mejoría. El paciente refirió tos seca, sobre todo en las noches, decaimiento marcado, más acentuado en horario vespertino, adelgazamiento progresivo en los últimos años, a pesar de alimentarseadecuadamente; además, percibió que sus brazos se “atrofiaban”. No refirió antecedente patológico personal alguno, ni hábitos tóxicos, solo antecedentes familiares de cardiopatía isquémica en la madre. Al interrogar a la familia, se supo que un hermano menor padecía los mismos síntomas, aunque nunca se había hecho ningún estudio.
Examen físico
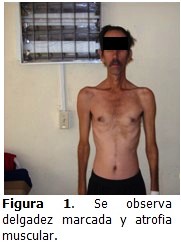
Al examinar al paciente, se constató la delgadez marcada, confacie delgada,calvicie, hipotrofia de los músculos maseteros, esternocleidomastoideos y de los músculos de las extremidades. (Figura 1). Peso: 52 kg.
Talla: 1,75 cm
Aparato respiratorio: murmullo vesicular disminuido en el tercio inferior de ambos hemitórax; estertores crepitantes finos a dicho nivel.
Aparatocardiovascular: ruidos cardiacos taquiarrítmicos. Ausencia de soplos. FC: 118x min. TA: 150/100mmHg.
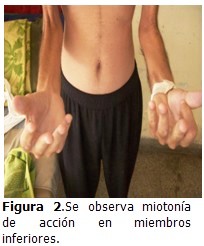
Aparato neurológico: signos de atrofia muscular y miotonía de acción. (Figura 2).
Fue tratado en base a una insuficiencia ventricular izquierda (edema pulmonar). Al mejorar los síntomas más apremiantes, se decidió su ingreso para estudio delestado cardiovascular y la distrofia muscular, constatada mediante fenómenos miotónicos.
Exámenes complementarios
En la radiografía de tórax se observó un aumento marcado del área cardiaca,con predominio del ventrículo izquierdo, engrosamiento hiliar de aspecto congestivo, así como lesiones congestivas hacia las bases.
Los exámenes de laboratorio mostraron resultados dentro de parámetros normales, incluida la creatina-fosfocinasa (CPK por sus siglas en inglés).
El electrocardiograma (ECG) mostrófibrilación auricular con respuesta ventricular acelerada.
En el ecocardiograma-doppler se observó un ventrículo izquierdo dilatado con hipocinesia y fracción de eyección del ventrículo izquierdo de un 35%. No se apreciaron lesiones valvulares.
La electromiografía evidenciómembrana muscular inestable, con actividad de inserción aumentada, abundante actividad de potenciales agudos positivos y fibrilares, descargas miotónicas de frecuencia variable y un patrón miopático en músculos proximales y distales de los miembros con unidades motoras polifásicas de baja amplitud. Fenómeno miotónicode frecuencia variable, espontáneo ysobre todo al percutir músculos o cambiar electrodos.
Se indicaron algunos fármacos y técnicas de fisioterapia para mejorar la calidad de vida del paciente. Fue remitido a la consulta de Clasificación de Trastornos Neuromusculares del Instituto Nacional de Neurología y Neurocirugía para su tratamiento y seguimiento.
DISCUSIÓN
La Federación Española de Enfermedades Neuromuscularesreconoce 12 tipos de enfermedades neuromusculares, entre las que se encuentra la distrofia miotónica de Steinert.3 La distrofia miotónica está compuesta por dos trastornos clínicos, con fenotipos superpuestos y defectos genéticos moleculares propios: el tipo 1 (DM1), que es la enfermedad clásica descrita originalmente por Steinert, y el tipo 2(DM2), llamado también miopatía miotónica proximal(PROMM). Ambas cursan con atrofia muscular, miotonía y cambios distróficos de tejidos no musculares. Las dos se transmiten de forma autosómica dominante.
Evidentemente el paciente presentaba manifestaciones cardiovasculares, y a la vez una distrofia muscular con fenómenos miotónicos, así como otras manifestaciones clínicas que hacían sospechar la entidad planteada, que aunque rara, es la más frecuente dentro de las distrofias musculares. El diagnóstico de DM1 o enfermedad de Steinert, se sospechó inicialmente debido a la existencia de un hermano con características similares, y a otros aspectos como la delgadez extrema, la calvicie y los trastornos del ritmo cardiaco; luego pudo sustentarse mediante los hallazgos en electrocardiograma, ecocardiograma y electromiografía.
La miotonía, contracción involuntaria mantenida de un grupo de músculos, es el síntoma neuromuscular cardinal de la distrofia miotónica. A menudo, los pacientes refieren rigidez y dificultad para soltar la mano, por ejemplo, después de un apretón de manos. La distrofia miotónica es hereditaria, como un rasgo autosómico dominante. Más del 95 % de los sujetos con distrofia miotónica tienen mutaciones en el gen que codifica la proteína quinasa de la distrofia miotónica (DMPK, siglas en inglés de myotonicdystrophyproteinkinase). En las personas sin la enfermedad, este gen contiene menos de 30 repeticiones de la secuencia CTG, mientras que en las afectadas gravemente puede haber varios miles de repeticiones. Por tanto, la distrofia miotónica entra en el grupo de trastornos asociados a expansiones por repetición de trinucleótidos. Como sucede en otros trastornos en los que existen mutaciones similares, la distrofia miotónica presenta el fenómeno de anticipación, caracterizado por el empeoramiento de las manifestaciones de la enfermedad con cada generación, debido al aumento del número de repeticiones de los trinucleótidos. La expansión por repeticiones CTG se localiza en la región no traducida 3´ del ácido ribonucleico mensajero (ARNm) de DMPK.4-8 En este paciente no se realizaron estudios genéticos, los cuales son útiles para confirmar el diagnóstico, pero no están al alcance de todos los centros hospitalarios, además de que son extremadamente costosos.1,5
La mayoría de los autores señala que la biopsia muscular no es necesaria. Si se practicara la biopsia muscular se observaría un músculo marcadamente comprometido por infiltración por tejido adiposo, con mayor afectación de algunos fascículos que otros. Las fibras que lo componen muestran marcada variación en su tamaño, evidenciándose pocas fibras con cambios degenerativos o regenerativos. El cambio más prominente es la internalización de los núcleos, presentes en un 68 % de las fibras, y en algunas la cantidad de núcleos es mucho más numerosa comparado con otras. También se observan algunas fibras en extremo atróficas, formando sacos de núcleos, así como el colágeno endomisialseveramente aumentado y cambios degenerativos de las fibras.1
En la DM1 la sintomatología muscular incluye debilidad de los músculos elevador de los párpados, maseteros y esternocleidomastoideo, los antebrazos, las manos y los músculos pretibiales. La afección de los maseteros causa un adelgazamiento de la mitad inferior de la cara, que junto a la ptosis palpebral y la calvicie precoz, confieren una facies característica (“cara de cuchillo”), como se muestra en el caso presentado. La debilidad de los músculos extensores de las muñecas, extensores de los dedos e intrínsecos de la mano, produce un déficit funcional. La debilidad de los flexores dorsales del tobillo puede dar lugar a pie péndulo. La afección palatina, faríngea y lingual produce disartria, voz nasal y problemas de deglución. Algunos pacientes presentan debilidad en el diafragma y los músculos intercostales, con la consecuente insuficiencia respiratoria.1-5,7 También se observan con frecuencia trastornos cardiacos; las anormalidades electrocardiográficas incluyen bloqueo auriculoventricular de primer grado, fibrilación auricular, aleteo auricular y otras alteraciones del sistema de conducción como bloqueo cardiaco completo. Pocas veces se observa insuficiencia cardiaca congestiva, pero puede ser consecuencia de la cardiopatía pulmonar que se establece por insuficiencia respiratoria, al ser tomados los músculos del tórax. El 65 % de los pacientes presenta alteraciones electrocardiográficas y con menor frecuencia, cuadros de miocardiopatías graves. A menudo, también se observa asociado prolapso de la válvula mitral.9-11
En un estudio de prevalencia de anomalías cardiacas en pacientes con DM1, los resultados de ECG realizados a 100 pacientes, informaron que la hipertrofia del ventrículo izquierdo (19,8 %), la dilatación del ventrículo izquierdo (18,6 %) y la disfunción sistólica del ventrículo izquierdo (14 %), se asociaron con mayor edad.El P-R>200 ms fue predictivo de anomalías regionales del movimiento de la pared; este y el QRS >120 ms se correlacionaron con anomalías regionales del movimiento de la pared y dilatación de la aurícula izquierda. La IC congestiva clínica se encontró en 1,8 % de los casos.
Otros signos acompañantes incluyen deficiencia intelectual, hipersomnia, cataratas subcapsulares posteriores, atrofia gonadal, resistencia a la insulina e hipomotilidad de esófago y colon. La miotonía suele preceder a la debilidad muscular en varios años y habitualmente aparece hacia los cinco años de edad. Siempre se produce tras activación muscular (miotonía de acción), generalmente voluntaria, aunque también puede aparecer por estimulación mecánica (miotonía por percusión) del músculo. La miotonía causa dificultades para liberar los objetos tras su sujeción firme con la mano. Los cambios distróficos de otros tejidos más frecuentes, son alopecia progresiva de inicio temprano, tanto en varones como en mujeres, y trastornos neuropsiquiátricos de leves a moderados. La muerte sobreviene por infecciones pulmonares, insuficiencia ventilatoria o insuficiencia cardiaca grave.1,4,6-10
En la literatura se recogen pocas experiencias con medicaciones específicas que hayan tenido éxito. Se citan buenos resultados con la administración de sulfato de dehidroepiandrosterona (DHEAS),precursor de las hormonas sexuales que conlleva a una mejoría de la fuerza muscular, de la miotonía, de los trastornos cardiacos y de la calidad de vida. También se sugiere la fenitoína para el control de la miotonia,2,4,5,7,8 que se prescribió en este caso como tratamiento específico para la insuficiencia cardíaca.La atención adecuada de estos casos requiere de la formación de equipos multidisciplinarios capaces de adaptarse a las particularidades de cada paciente, ya que la enfermedad evoluciona de forma progresiva, con complicaciones, discapacidad, repercusión psicológica en el individuo y en la familia.
Aunque se trate de una enfermedad rara, ante la observación de un paciente con atrofia muscular, miotonía y manifestaciones cardiacas, entre otras, debe sospecharse la posibilidad de la enfermedad descrita por Steinert; deben buscarse los otros elementos que conforman su expresión clínica, a través de los diferentes medios diagnósticos ya descritos. En este caso, el interrogatorio adecuado, el examen exhaustivo y la observación del paciente, permitieron sospechar el diagnóstico y orientar los estudios necesarios.
